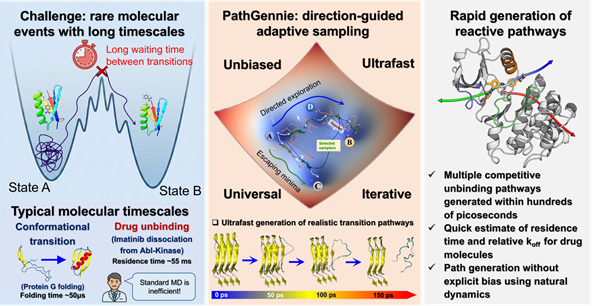
Scientists have developed a novel computational framework called PathGennie that promises to accelerate drug discovery by efficiently simulating rare molecular events — a key bottleneck in identifying and optimising therapeutic compounds.
Developed by researchers and published in the Journal of Chemical Theory and Computation, PathGennie is an open-source software tool that predicts how candidate drug molecules unbind from their protein targets without relying on the artificial steering forces typical of standard simulation techniques. This gives more realistic insights into molecular interactions crucial for drug design.
By reducing the time and computational effort needed to model complex molecular processes, PathGennie could significantly fast-track computer-aided drug discovery (CADD), enabling researchers to explore candidate molecules more rapidly and with greater accuracy.
Experts say such computational advances are important as drug development timelines remain long and costly — often taking years and substantial investment to identify viable drug candidates.
Significance: The new method offers a practical, accelerated approach to simulate molecular behaviour, which may help streamline early-stage discovery, reduce costs, and improve the selection of promising therapeutic leads well before laboratory testing.